Mapping Yeast Traits: A Closer Look at Genes in Each Cell
Jim Crocker
31st July, 2025



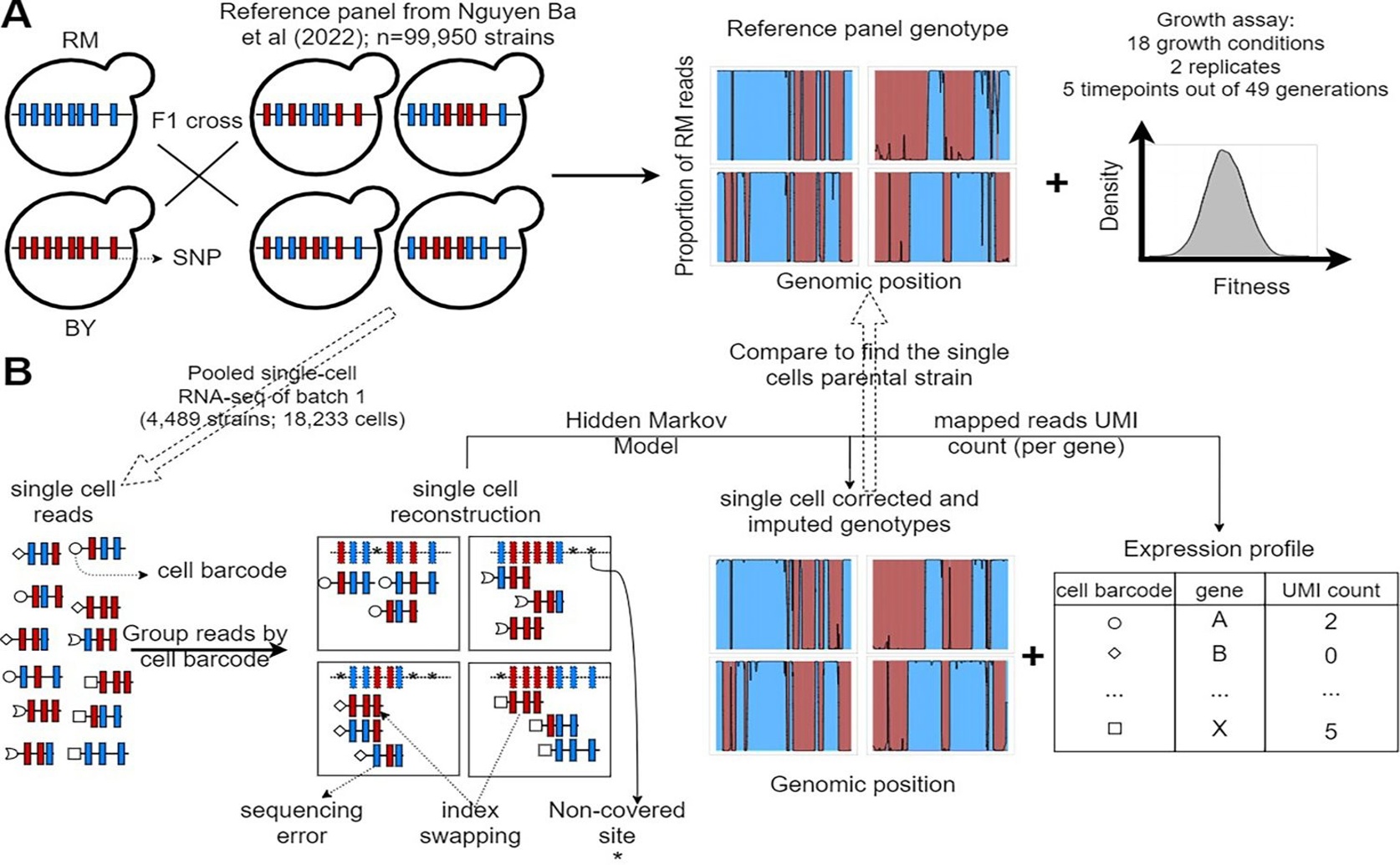
By leveraging a reference panel of yeast (Saccharomyces cerevisiae) segregants characterized by bulk sequencing (a), this study validates a high-throughput single-cell RNA sequencing approach (b) that accurately captures linked genotype and transcriptomic profiles to refine the resolution of the genotype–phenotype map.
Key Findings
- Researchers at the University of Toronto used a new single-cell RNA sequencing method on yeast to better map genetic differences to traits
- Their large-scale study revealed that genetic elements far from a gene (trans-acting) largely control gene activity more than nearby ones (cis-acting)
- This improved method also identified new gene regulation hotspots and highly heritable gene functions, confirming gene expression significantly shapes traits
References
Main Study
1) Refining the resolution of the yeast genotype–phenotype map using single-cell RNA-sequencing
Published 28th July, 2025
https://doi.org/10.7554/eLife.93906
Related Studies
2) Finding the sources of missing heritability in a yeast cross.
3) Genetics of trans-regulatory variation in gene expression.
4) A high-definition view of functional genetic variation from natural yeast genomes.
5) Rare variants contribute disproportionately to quantitative trait variation in yeast.
Related Articles
 17th June, 2025 | Jim Crocker
17th June, 2025 | Jim Crocker