DNA Tags in Single Cells Reveal Cell History, Gene Activity, and Growth
Jenn Hoskins
11th July, 2025



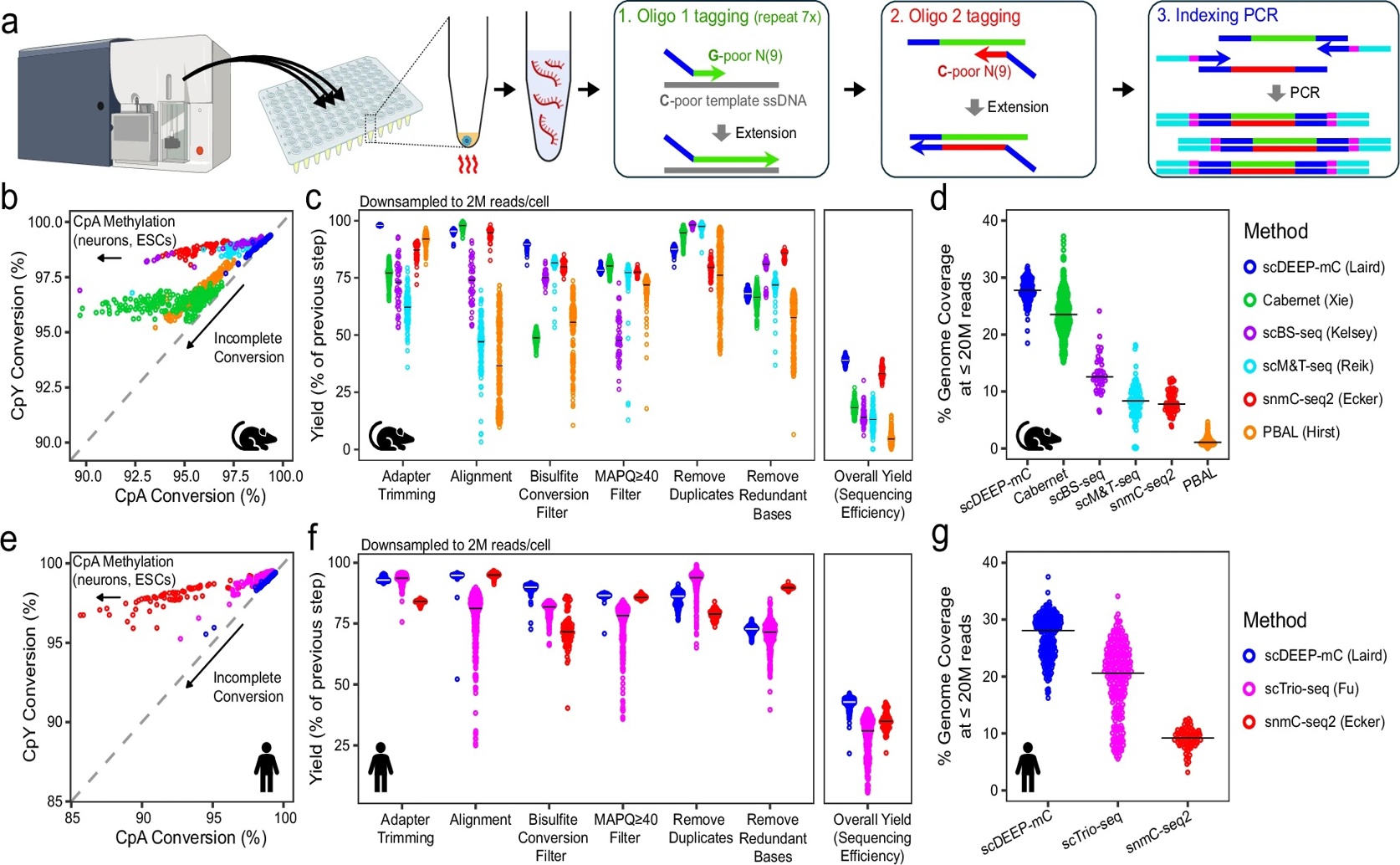
The scDEEP-mC library preparation workflow (a) outperforms existing single-cell whole-genome bisulfite sequencing methods in both mouse and human cells, demonstrating consistent bisulfite conversion (b, e), the highest sequencing efficiency (c, f), and superior genomic coverage (d, g).
Key Findings
- This new method, scDEEP-mC, developed at Van Andel Institute, vastly improves mapping DNA methylation patterns in individual cells with high detail
- This advanced method allows scientists to precisely identify different cell types and track how DNA tags change as cells divide or during processes like X-inactivation
References
Main Study
1) High-coverage allele-resolved single-cell DNA methylation profiling reveals cell lineage, X-inactivation state, and replication dynamics
Published 8th July, 2025
https://doi.org/10.1038/s41467-025-61589-1
Related Studies
2) DNA methylation in mammalian development and disease.
3) The Epigenetic Hallmarks of Cancer.
4) High-Resolution Single-Cell DNA Methylation Measurements Reveal Epigenetically Distinct Hematopoietic Stem Cell Subpopulations.
5) Single-cell multiomics sequencing and analyses of human colorectal cancer.
Related Articles
 11th April, 2025 | Jim Crocker
11th April, 2025 | Jim Crocker