Predicting Protein Interactions and Their Role in Phosphorylation
Jenn Hoskins
6th March, 2025



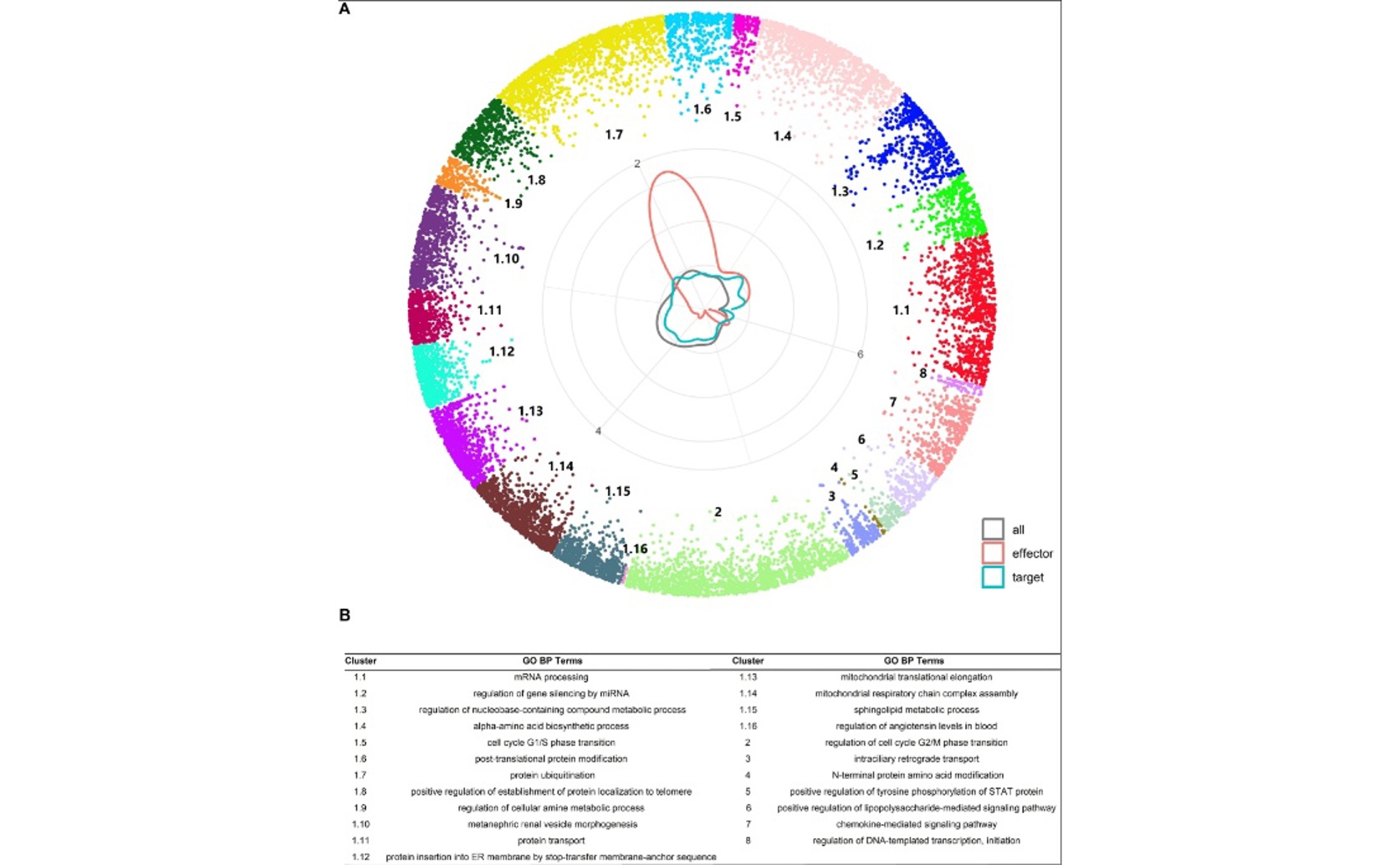
This figure demonstrates that proteins involved in experimentally validated phosphorylation and dephosphorylation interactions (effectors and targets) occupy distinct, functionally enriched angular clusters in the hyperbolic human protein interaction network, indicating that hyperbolic embedding captures biologically meaningful organization relevant to predicting PTM-directed PPIs.
Key Findings
- *Mainz researchers mapped human protein interactions using advanced geometry to understand cell functions.*
- *They applied machine learning to predict key protein interactions linked to various diseases.*
- *The study identified specific protein disruptions in a neurodegenerative disease, paving the way for new treatments.*
References
Main Study
1) Prediction of protein interactions with function in protein (de-)phosphorylation
Published 3rd March, 2025
https://doi.org/10.1371/journal.pone.0319084
Related Studies
2) Towards a proteome-scale map of the human protein-protein interaction network.
Journal: Nature, Issue: Vol 437, Issue 7062, Oct 2005
3) A human interactome in three quantitative dimensions organized by stoichiometries and abundances.
4) Protein-protein interaction networks: unraveling the wiring of molecular machines within the cell.
5) Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery.
Related Articles
 11th February, 2025 | Jenn Hoskins
11th February, 2025 | Jenn Hoskins