How Proteins Change Shape: AI And X-Ray Views
Jenn Hoskins
29th June, 2025



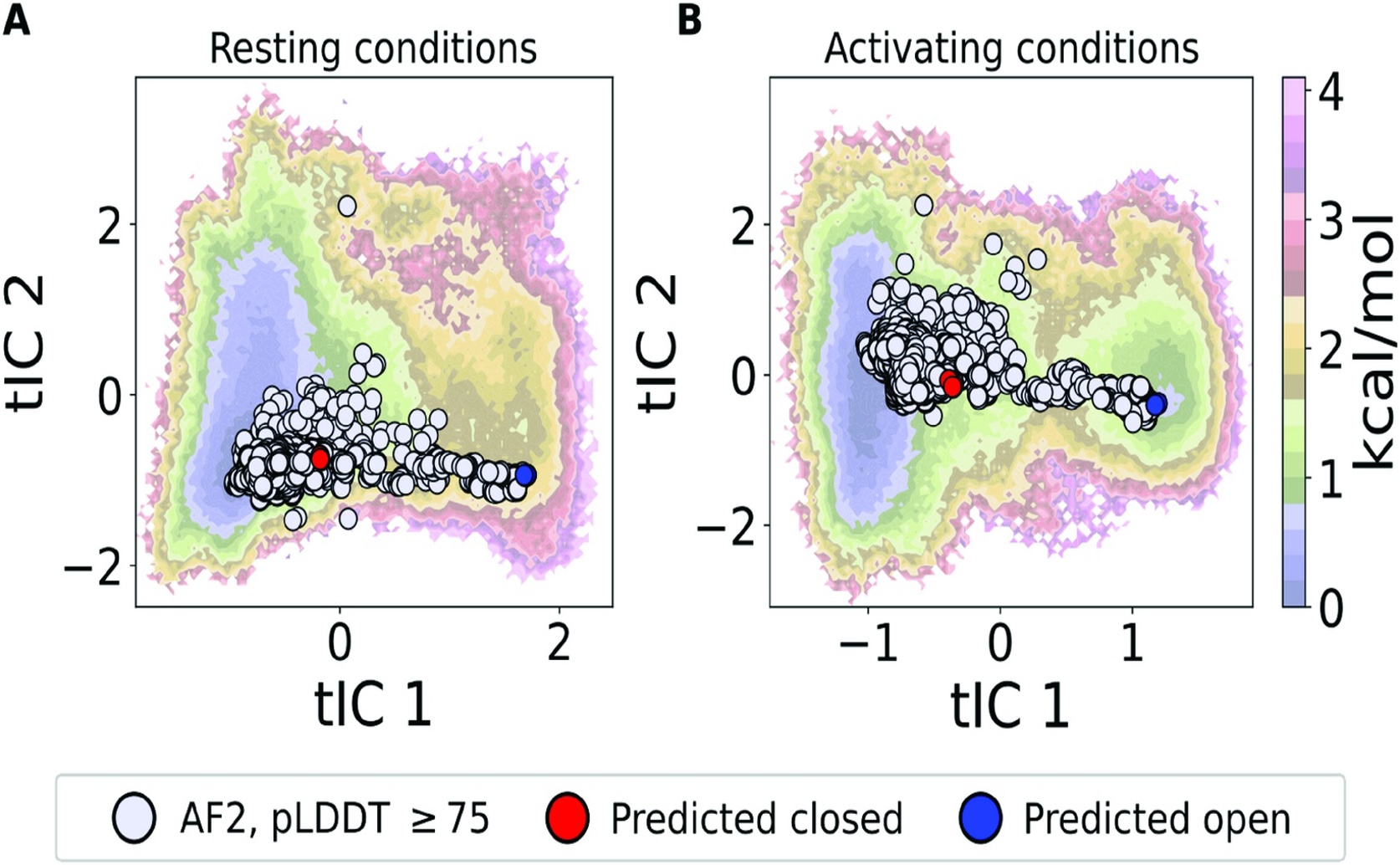
Projections of the AlphaFold-generated ensemble onto free energy landscapes derived from molecular dynamics simulations confirm that the predicted structures align with the energetic minima and transition pathways of the GLIC protein under deprotonated (a) and protonated (b) conditions.
Key Findings
- Researchers at Science for Life Laboratory and the University of Kansas developed a new method combining AI (AlphaFold2) and SANS to rapidly map protein shape changes, focusing on the GLIC ion channel
- This method accurately identified the GLIC channel's distinct "closed" and "open" states and precisely measured their populations under different conditions
- Crucially, it mapped the entire dynamic pathway of shape transitions, including fleeting intermediate forms, thousands of times faster than traditional simulations
References
Main Study
1) Resolving the conformational ensemble of a membrane protein by integrating small-angle scattering with AlphaFold
Published 27th June, 2025
https://doi.org/10.1371/journal.pcbi.1013187
Related Studies
2) Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins.
3) Conformation-specific Synthetic Antibodies Discriminate Multiple Functional States of the Ion Channel CorA.
4) Cryo-EM structures of prokaryotic ligand-gated ion channel GLIC provide insights into gating in a lipid environment.
Related Articles
 17th June, 2025 | Jim Crocker
17th June, 2025 | Jim Crocker