Can We Spot Adaptation in Tiny Samples and Chance?
Jenn Hoskins
25th June, 2025



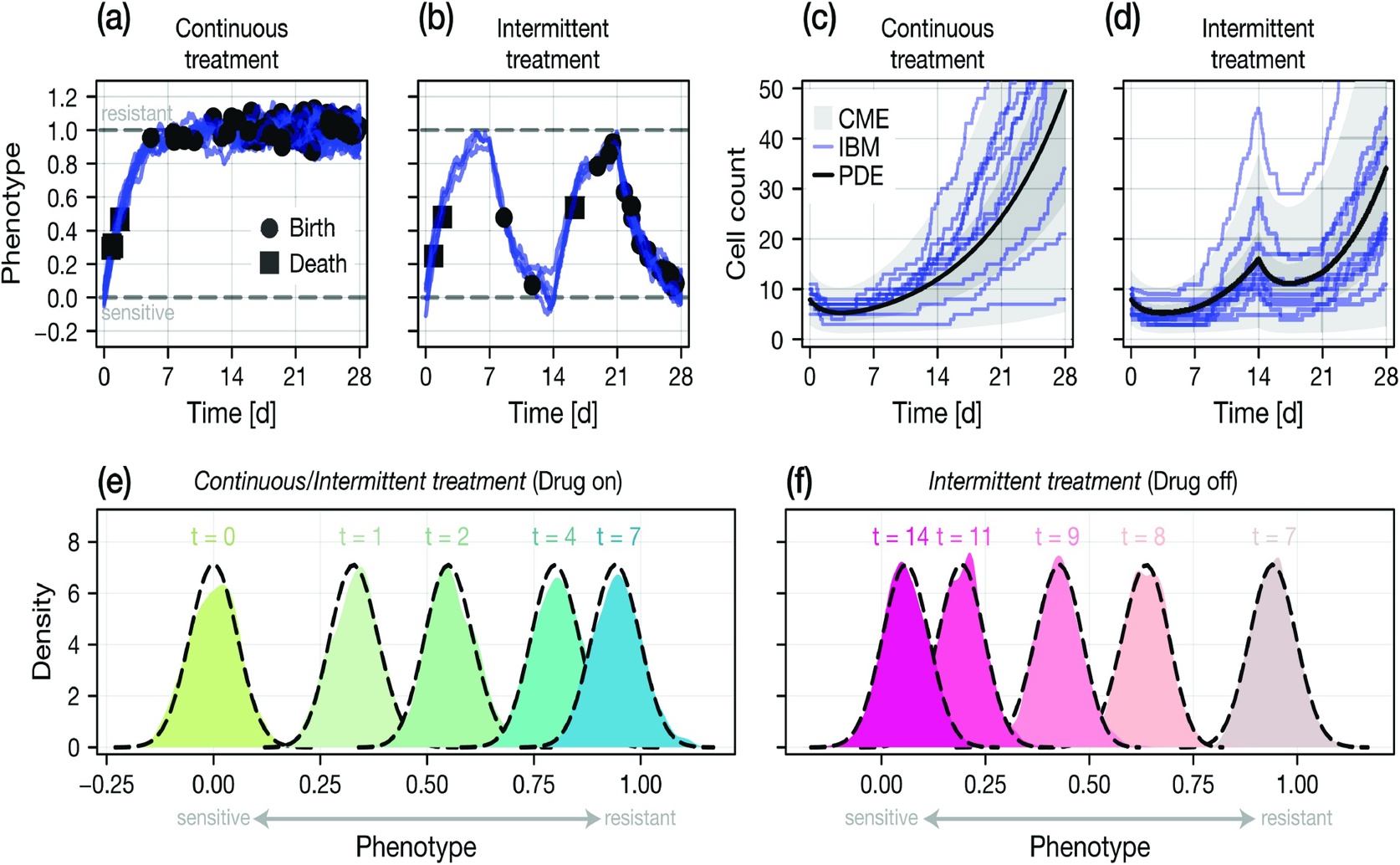
The study’s mathematical models are validated by accurately capturing the average phenotypic distribution (e–f) and the full stochastic cell count dynamics (c–d) observed in the underlying individual-based simulations of continuous (a, c) and intermittent (b, d) drug treatment.
Key Findings
- The study from International Universities & Research Centers found that standard population-level cell count data cannot reliably show how much individual cancer cells differ in their drug resistance
- Furthermore, these common cell count measurements also fail to distinguish if drug resistance emerges as distinct cell types or as a gradual shift across a spectrum of resistance
- To truly understand this cellular adaptability, more detailed single-cell data, like observing individual cell growth and death events, are needed
References
Main Study
1) Identifiability of phenotypic adaptation from low-cell-count experiments and a stochastic model
Published 24th June, 2025
https://doi.org/10.1371/journal.pcbi.1013202
Related Studies
2) The great escape: tumour cell plasticity in resistance to targeted therapy.
3) Therapy resistance: opportunities created by adaptive responses to targeted therapies in cancer.
4) Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance.
Related Articles
 9th June, 2025 | Jenn Hoskins
9th June, 2025 | Jenn Hoskins