AI Predicts Disease Links In Our Genes
Greg Howard
20th June, 2025



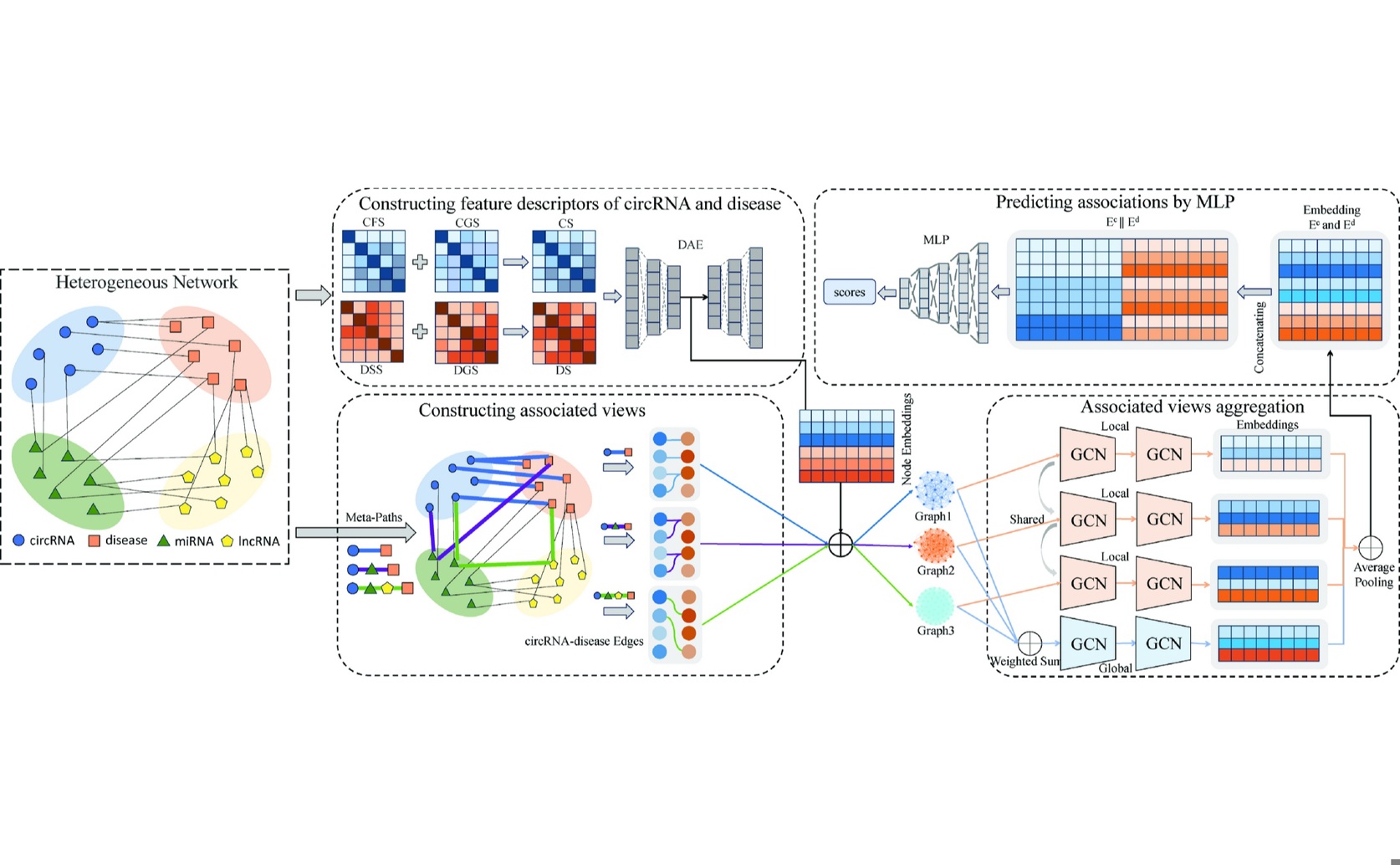
The MVHGCN workflow predicts circRNA-disease associations by integrating diverse data sources into a heterogeneous graph, from which multiple relational views are extracted via meta-paths and aggregated using a graph convolutional network.
Key Findings
- Researchers from Chinese universities developed MVHGCN, a new AI tool, to accurately predict links between circular RNAs and diseases, overcoming data sparsity issues
- This method builds a comprehensive network of biological data and uses advanced AI to find hidden, indirect connections between circRNAs and diseases
- MVHGCN significantly outperforms previous methods, offering a powerful new way to accelerate disease research and develop new treatments
References
Main Study
1) MVHGCN: Predicting circRNA-disease associations with multi-view heterogeneous graph convolutional neural networks
Published 19th June, 2025
https://doi.org/10.1371/journal.pcbi.1013225
Related Studies
2) Circular RNAs are a large class of animal RNAs with regulatory potency.
3) DGCLCMI: a deep graph collaboration learning method to predict circRNA-miRNA interactions.
4) Prediction of CircRNA-Disease Associations Using KATZ Model Based on Heterogeneous Networks.
5) CRBPSA: CircRNA-RBP interaction sites identification using sequence structural attention model.
Related Articles
 16th July, 2024 | Jim Crocker
16th July, 2024 | Jim Crocker