Flexible Protein Sequences Impact Structure
Jenn Hoskins
23rd April, 2025



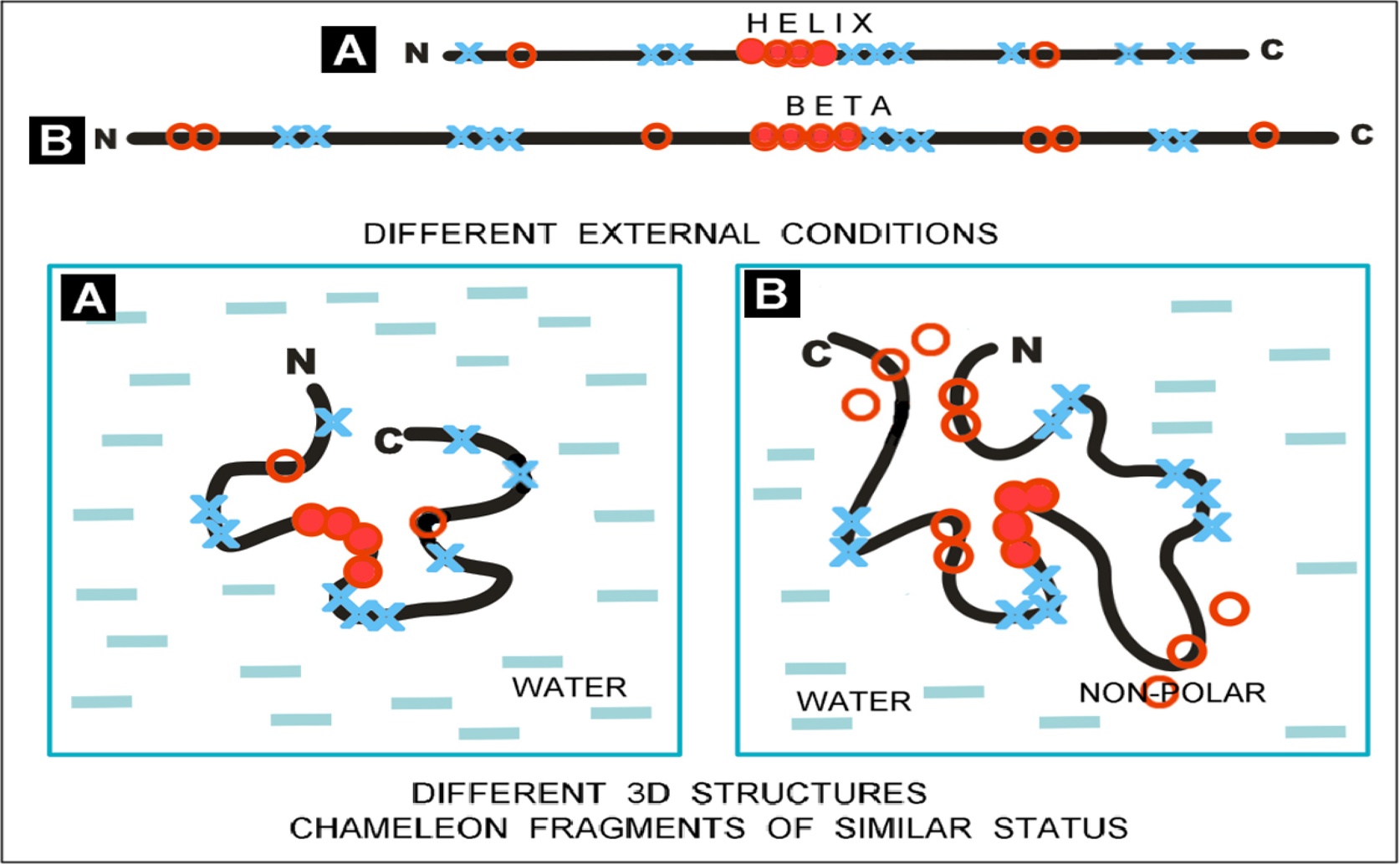
This conceptual model demonstrates that the polarity of the local environment dictates whether a chameleon sequence folds into a helical or beta-sheet structure (a, b), illustrating that secondary structure formation is subordinate to achieving a functionally compatible overall protein conformation.
Key Findings
- Researchers at Silesian University found that the surrounding hydrophobic environment primarily guides how proteins fold, not just their amino acid sequences
- This helps explain how "chameleon sequences" can adopt different shapes, improving our understanding of diseases caused by misfolded proteins and aiding drug development
- The study's insights could lead to better predictions and treatments for conditions like Alzheimer's by targeting protein folding processes
References
Main Study
1) Chameleon sequences—Structural effects
Published 22nd April, 2025
https://doi.org/10.1371/journal.pone.0315901
Related Studies
2) Analysis of protein chameleon sequence characteristics.
Journal: Bioinformation, Issue: Vol 3, Issue 9, May 2009
3) Dependence of Protein Structure on Environment: FOD Model Applied to Membrane Proteins.
4) Model of the external force field for the protein folding process-the role of prefoldin.
5) Ab initio protein structure prediction: the necessary presence of external force field as it is delivered by Hsp40 chaperone.
Related Articles
 11th April, 2025 | Jim Crocker
11th April, 2025 | Jim Crocker